MXene作为一种新型二维过渡金属碳/氮化物,在固体润滑领域展现出巨大潜力。氮掺杂被证实可有效调控MXene的电子结构和界面行为,但其原子尺度摩擦机理及掺杂位点依赖性尚不明确。密度泛函理论(DFT)计算是获取原子尺度材料本征参数的精确工具,但其高昂的计算成本使得对多种MXene材料及多个掺杂位点的系统研究极为耗时,导致该领域长期缺乏规范、完整的高通量数据集,严重制约了高性能氮掺杂MXene润滑涂层的理性筛选与设计。
近日,兰州交通大学材料科学与工程学院与中国科学院兰州化学物理研究所科学数据中心合作,采用自主搭建的固体界面摩擦性能第一性原理高通量计算平台LICP-FPHTC-Platform(摩擦学学报42(2022)493;软件著作权2021SR1172689),系统计算了5种MXene材料(Nb-C、V-C、Sc-C、Ta-C、Mo-C)在4种不同氮掺杂位点下的电子、力学、热力学及摩擦学性质,构建了一个包含209个结构的高质量DFT数据集(图1)。
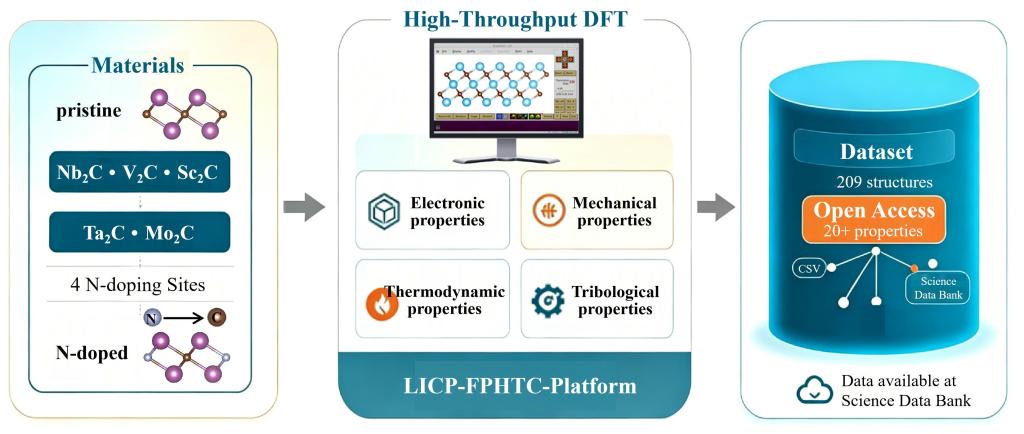
图1氮掺杂MXene高通量DFT数据集构建流程示意图
该数据集系统记录了晶格参数、层间接触面积、结合能、形成能、杨氏模量、剪切模量、泊松比、滑动能垒及摩擦力等20余项关键参数,并通过与文献对比验证了计算方法的可靠性。该数据资源不仅为理解氮掺杂MXene的原子尺度润滑机理提供了基础数据支撑,也为利用机器学习方法高通量筛选低摩擦、高可靠性的N掺杂MXene涂层建立了重要的基准数据库。
相关成果以“N-doped MXenes for tribological applications: A high-throughput DFT dataset”为题发表在Nature子刊《Scientific Data》期刊(https://doi.org/10.1038/s41597-026-07236-w)。兰州交通大学硕士生徐明阳为该论文的第一作者,兰州交通大学耿中荣教授与兰州化物所高瑜助理研究员为该论文的共同通讯作者。
该研究工作得到了中国科学院战略性先导科技专项、国家自然科学基金、中国科学院“西部之光-西部交叉团队”项目、甘肃省重点研发计划及兰州化物所“十五五”重点培育项目的支持。
(文图/耿中荣 初审/郭鹏 复审/褚克 终审/吴刊选 校对/张一 来源/材料学院)







